
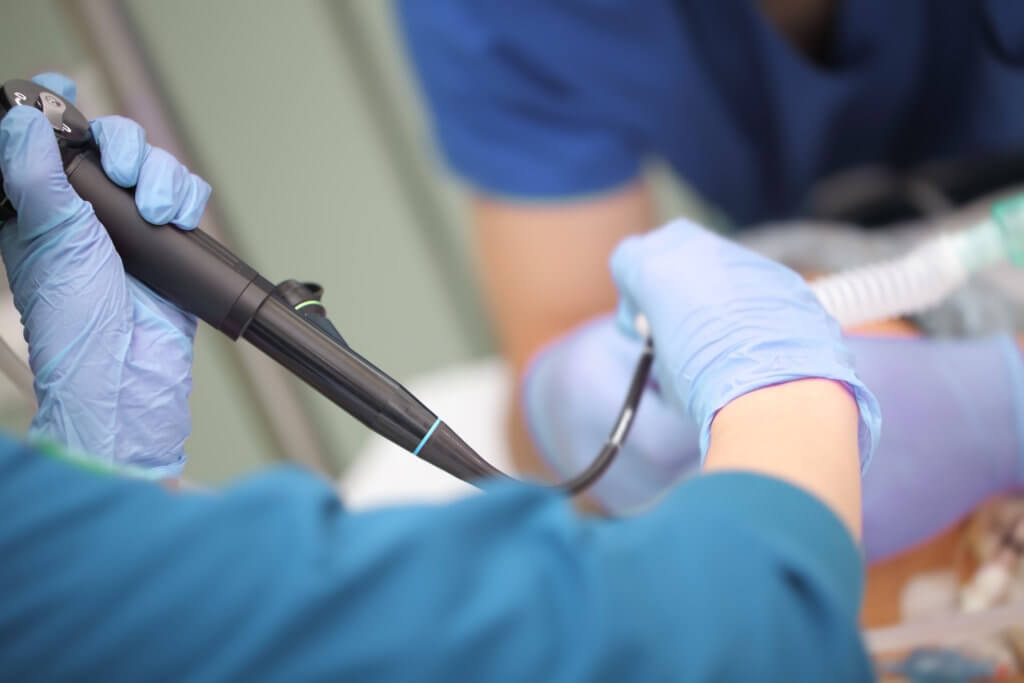
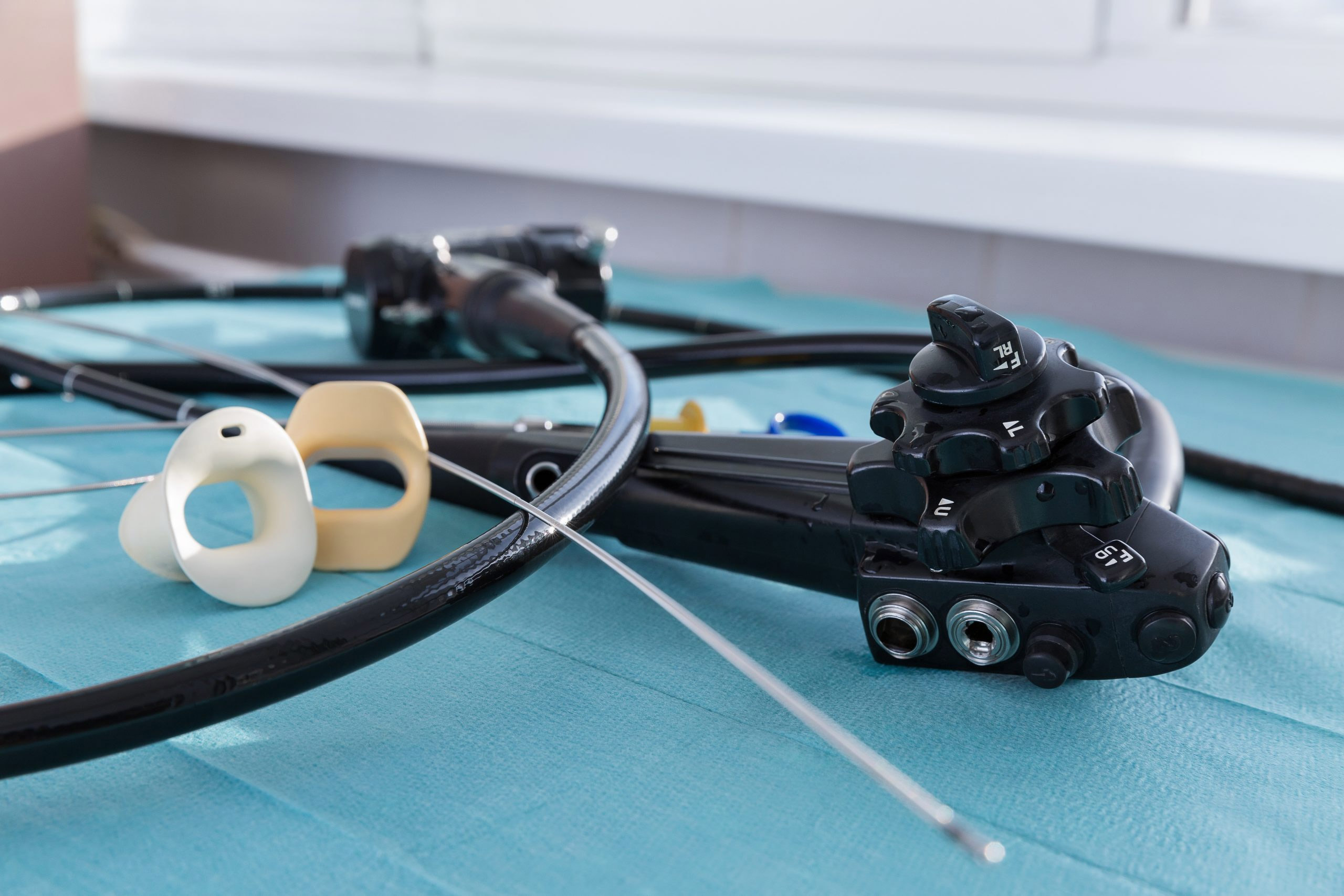
The U.S. Food and Drug Administration is investigating “numerous” medical device reports (MDRs) describing patient infections and other possible contamination issues possibly associated with reprocessed urological endoscopes.
These endoscopes include cystoscopes, ureteroscopes, and cystourethroscopes.
Between Jan. 1, 2017 and Feb. 20, 2021, the FDA received more than 450 MDRs describing post-procedure patient infections or other potential contamination issues involving reprocessing these endoscopes, the agency said in a letter to healthcare providers issued April 1. The FDA says it is investigating potential causes and contributing factors associated with the reported infections and contamination issues.
Some reports have indicated issues with inadequate reprocessing or maintenance. Other potential issues the FDA is evaluating include device design and reprocessing instructions on labeling.
The FDA, still early in its evaluation, believes the risk of infection is low based on available data. Two manufacturers were cited in reports that named the maker of the devices in question. However, the agency “has not concluded that such risks are limited to a particular manufacturers’ devices nor that any specific manufacturer or brand of these devices is associated with higher risks than others.”
The reports include three deaths which occurred outside of the U.S. The patients developed Pseudomonas aeruginosa infections after procedures.
One patient death involved a cystoscope, which the report said did not pass a leak test — a preventive maintenance process required to ensure the device is not damaged. Failure to ascertain that the scope was damaged could be a source of infection.
The two other reported deaths were associated with the use of a forceps/irrigation plug, an accessory component used to control water flow and enable access to the working channel of the endoscope.
“It is unknown whether or to what degree the reported infections contributed to the patient deaths, and patient co-morbidities may have been a factor,” the FDA said in its statement. “MDRs are not, by themselves, definitive evidence of a faulty or defective medical device, and cannot be used to establish or compare rates of event occurrence.”
The FDA’s announcement includes the following recommendations for healthcare providers:
The agency encourages healthcare providers to report any suspected adverse events experienced with cystoscopes, ureteroscopes, and cystourethroscopes.
In August 2020, the FDA updated its recommendation that duodenoscope manufacturers and healthcare facilities transition to duodenoscopes with disposable components and scopes that are fully disposable. The safety communication from the FDA followed a prior one issued in August 2019 calling for manufacturers and healthcare organizations to move to duodenoscopes that are partially or fully single-use.


A Learning Center by Ambu USA


